Antony N. Daviesa,b
aStrategic Research Group – Measurement and Analytical Science, Akzo Nobel, Deventer, the Netherlands
bSERC, Sustainable Environment Research Centre, Faculty of Computing, Engineering and Science, University of South Wales, UK
Tandem mass spectrometry1 has become a tool of choice for the identification of trace levels of compounds, especially in data-rich environments such as metabolomics and increasingly for screening in toxicological or forensic investigations. The benefits of adopting such methods lie in the low detection limits and the ability to extract quantitative information from samples with impossibly difficult to impossible clean-up options with multiple analytes co-eluting from the chromatography stage. Most of these techniques rely heavily on prior knowledge of the ionisation profiles of the analytes being studied. Where the metabolites are potentially unknown, it would be possible to resort to database searching of the signals observed, however, here, as in many areas of spectroscopy, the lack of comprehensive reference database coverage means that the spectroscopist has to resort to other methods to identify the newly detected compounds. This article looks at the background to the problem and a novel solution—CSI:FingerID—which allows for highly sensitive tandem mass spectrometry (MS/MS) data to be used to identify unknown analytes from common molecular structure databases where reference spectroscopic data is unavailable.
Tandem mass spectrometry
So, what are we talking about? Tandem mass spectrometry has been available for many years and deployed in many fields where identification and quantification of analytes is not possible by a single mass detector coupled to a chromatography sample preparation step. There are a few common main instrumental configurations depending on experiments to be performed. Figure 1 from Rouessac and Rouessac2 provides a common instrumental schematic for an instrument capable of MS/MS experiments using three quadrupoles.

Figure 1. A schematic of a simple triple quadrupole tandem mass spectrometer. The middle quadrupole is used as a collision chamber. These instruments can conduct all three common types of MS/MS analysis shown in Figure 2. For the untargetted metabolomics experiment a much higher mass resolution would be required such as from a Q-ToF or Orbitrap. Reproduced from Reference 2 with permission. © John Wiley & Sons.

Figure 2. Three of the most common MS/MS experiments following the initial ionisation step. 1) Scanning the first quadrupole whilst keeping the second fixed on a specific product ion; 2) keeping the first quadruple fixed on a specific precursor ion whilst scanning the second to obtain a full product ion spectrum; or 3) keeping both quadrupoles fixed to only allow a specific precursor and product ion pair to be detected.
Targeted vs untargeted tandem mass spectrometry
Clearly, some of the experiments indicated in Figure 2 require prior knowledge of exactly which analytes you wish to detect and their primary and secondary ionising fingerprints. These “targeted” analyses can be extremely sensitive and selective for specific analytes such as in screening for banned substances in a forensic environment or toxicological screening. This is less useful in areas such as metabolomics studies of new biological samples, where the vast majority of the metabolites observed are initially unknown. Gary Patti and co-workers3 have a simple to understand figure in their paper showing how the targeted and untargeted approaches play different roles in metabolomics—targeted answering questions around specific levels of known metabolites in samples with untargeted looking at the global metabolic profile of the sample and showing the complex MS/MS picture being the basis for subsequent measurement of MS/MS standards to support substance identification.
CSI:FingerID supporting interpretation of untargeted MS/MS analyses
Sebastian Böcker from the Friedrich Schiller University in Jena, Germany, with his colleagues Kai Dührkop and Marvin Meusel along with Juho Rousu and Huibin Shen from the Institute for Information Technology at Aalto University in Espoo, Finland, looked at the problem of structural elucidation of the results of untargeted tandem mass spectrometry experiments caused by the lack of good quality reference MS/MS data. The approach described by Patti—although eventually providing good results—relies on the availability of reference standard materials for the individual metabolites if it is not to be time consuming. The failure of the community to generate large, good quality reference data collections is compounded by the enormous improvements in instrumentation and sample preparation techniques delivered by the instrument manufacturers. It does provide, however, an excellent hunting ground for innovative bioinformaticians.
The current CSI:FingerID has been built on experiences gained from previous work4 in this area and has three main sections. Before looking at the unknown spectra, the system is trained in a learning phase—calculating fragmentation trees from known reference spectra, fragmentation tree similarities as well as PubChem (CACTVS)5 and Klekota–Roth fingerprints6 (see Table 1). In the current version, each molecular property is predicted by an individual “support vector machine” (SVM) which together make up the nerve centre for the approach. The SVMs yield probabilities for the presence or absence of a particular molecular property in any given compound based on the MS/MS spectral data.
| Data source | Number | Use |
| GNPS Public Spectral Libraries6 | 4138 | Training spectra |
| 3868 | Validation spectra | |
| MassHunter Forensics/Toxicology PCDL library7 | 2120 | Training spectra |
| 2055 | Validation spectra | |
| MassBank8 | 625 | Validation spectra |
| PubChem5 | 52,926,405 40,805,940 | Compounds Structures |
| PubChem filtered biodatabase9 | ~300,000 268,633 | Compounds Structures |
With the system trained, it is now possible to move to the actual analysis of new data. The system takes one or more MS/MS spectra, the unknown analyte to be identified and calculates the similarities shown in the unknown data set against the MS/MS spectra and fragmentation trees in the training set. The SVMs then predict the presence or absence of all molecular properties for the unknown compound providing a probability fingerprint.
This fingerprint can then be used as a search criterion against the much bigger molecular structure databases such as PubChem. Each potential solution chemical structure has its calculated fingerprint scored against the unknown to provide a hit-list to the user.
CSI:FingerID validation and comparison against existing methods
For the validation of this approach against existing prediction algorithms, the authors took the normal step of ensuring that none of the compounds used for testing were in the phase one training data sets. Ten-fold validation was carried out ensuring that no repeat batches contained the same structures. The authors provide a lot of statistics for different tests they have carried out against earlier versions of their software and other approaches for this task. Suffice to say that the latest version has yielded a 2.5-times improvement in predicting the correct molecular structure for the MS/MS unknown to 34.4% in the first place in their results list against the PubChem database and an impressive 63.5% of the unknown MS/MS spectra had their correct structures predicted in the top five hits. This is a very strong result.9
As phase one uses a machine learning stage which includes not only fragmentation tree information but also reference, known MS/MS data sets to train the system, the authors have also attempted to quantify how this size of this training data set alters the prediction capacity for the system as a whole. By reducing the amount of reference data, they could degrade their prediction capabilities, and carrying out a series of experiments came to the unsurprising conclusion that, with the limited amount of available good quality reference MS/MS data, they were far from saturating the prediction capability of their algorithms, the quality roughly increasing by around one percentage point for each additional 400 reference data sets added to the system.
So—a clear call for more data to be made available—again!
Try it out!
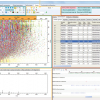
CSI:FingerID is available for you to try out at http://www.csi-fingerid.org/. There is a simple user interface (Figures 3–5) and what I really like the ability to run through the sequences of steps using demo data which quickly allows the user to see what they should be inputting.



Figures 3-5. Top: First stage—input your data. Middle: Second stage—the prediction engine works on the possible molecular formulae. Bottom: Third stage—Potential compounds are identified from the PubChem data collection.
References
- IUPAC, Compendium of Chemical Terminology, 2nd Edn (the “Gold Book”), Compiled by A.D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997); M. Nic, J. Jirat and B. Kosata, XML on-line corrected version, updates compiled by A. Jenkins (2006). doi: http://dx.doi.org/10.1351/goldbook
- F. Rouessac and A. Rouessac, Chemical Analysis, Modern Instrumentation Methods and Techniques, 2nd Edn. John Wiley & Sons Ltd, Fig. 16.5, p. 401 (2007). ISBN 978-0-470-85903-2
- G.J. Patti, O. Yanes and G. Siuzdak, “Innovation: Metabolomics: the apogee of the omics trilogy”, Nat. Rev. Mol. Cell Biol. 13, 263–269 (2012). doi: http://dx.doi.org/10.1038/nrm3314
- S. Böcker and F. Rasche, “Towards de novo identification of metabolites by analysing tandem mass spectra”, Bioinformatics 24(16), i49–i55 (2008). doi: http://dx.doi.org/10.1093/bioinformatics/btn270
- https://pubchem.ncbi.nlm.nih.gov/
- https://gnps.ucsd.edu/ProteoSAFe/libraries.jsp
- Agilent Technologies Inc.
- H. Horai et al., “MASSBANK: a public repository for sharing mass spectral data for life sciences”, J. Mass Spectrom. 45(7), 703–714 (2010). doi: http://dx.doi.org/10.1002/jms.1777
- K. Dührkop, H. Shen, M. Meusel, J. Rousu and S. Böcker, “Searching molecular structure databases with tandem mass spectra using CSI:FingerID”, Proc. Natl. Acad. Sci. USA in press. doi: http://dx.doi.org/10.1073/pnas.1509788112